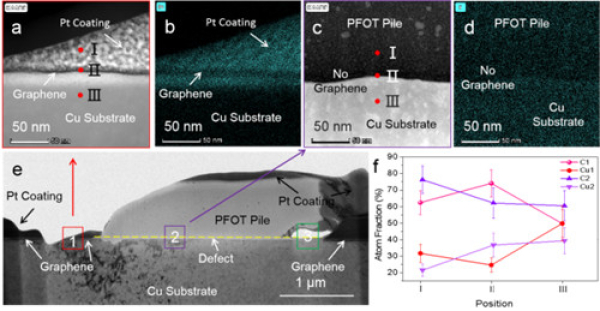
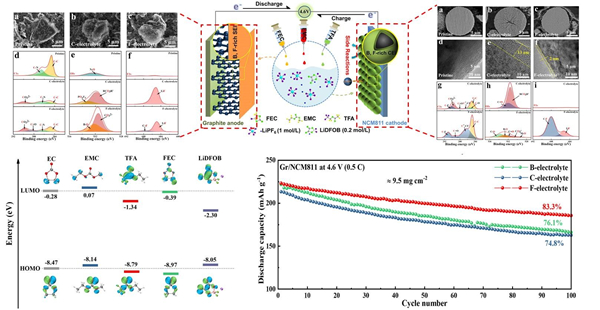
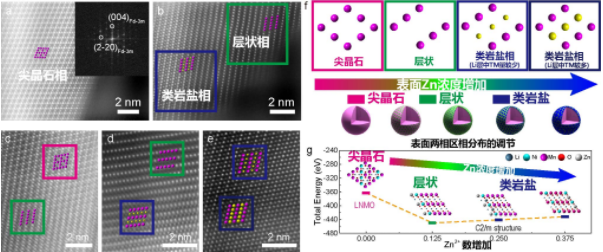
近年来,水系二次电池因为安全、绿色和低成本受到关注,但受限于水系电解液电化学窗口窄,水系二次电池存在输出电压低,能量密度低,自放电高以及循环寿命差等问题,难以推广应用。为了实现宽电位水系电解液,超高盐浓度Water-in-salt电解液被提出,通过提高盐浓度实现溶剂-离子相互作用调制降低水的电化学活性和实现SEI离子导体钝化膜,将水系电解液电化学窗口拓宽至3V及以上(Science 350,6263,2015;JACS 139,51,2017)。但是,伴随着电解液盐浓度的不断提升,电解液粘度急速上升,电导率快速下降,固液转化温度趋于室温,导致超高盐浓度WIS电解液动力学性能和低温性能变差、电解液成本升高等问题。因此,如何在宽电位电解液中实现优异的动力学性能和低温性能和低成本,成为水系电解液和水系二次电池面临的关键技术挑战。
近期,中国科学院物理研究所/北京凝聚态物理国家研究中心怀柔研究部、清洁能源实验室博士岳金明在研究员索鎏敏指导下,在研究不同溶解气体对SEI膜形成和稳定影响过程中(图1、2),发现LiTFSI水溶液与CO2之间存在特殊的强相互作用,该作用可以促使形成富CO2电解液体系,并在随后的电化学过程中在负极还原形成Li2CO3,实现SEI膜动力学保护,从而实现电解液电化学窗口的拓宽。进一步研究发现,这种强相互作用对盐的浓度具有依赖性,并呈现先增后减的趋势,在低盐浓度5 m LiTFSI(Salt-in-Water,SIW)时最强(图4)。与传统超高盐浓度WIS(21 LiTFSI-H2O)电解液相比,CO2-SIW电解液不但具有相当的宽电化学窗口,而且离子电导率是前者的10倍,粘度降低了一个数量级,固-液转变温度下降了50度(为-40℃), 成本降低了44 %(图3),这些物理化学变化为未来设计低成本、高性能、高电压水系电池提供更大空间。
研究进一步提出采用富CO2的低盐浓度电解液 (5 m LiTFSI)(CO2-SIW)来实现高电压水系锂离子电池 (> 2 V),采用该类电解液的2V水系锂离子电池(LiMn2O4-Mo6S8)表现出优良的电化学性能,并且循环过后的Mo6S8负极表面形成了有效的SEI钝化膜(图5)。得益于低盐浓度电导率高、粘度低、液相点温度低等优势,水系锂离子全电池动力学性能和低温性能得到了大幅提升,主要表现为在超厚和高载量电极(334 um,40 mg/cm2)以及-40 ℃低温条件下可以稳定高效工作,这是原有超高盐浓度WIS电解液无法实现的(图6)。
该工作研究成果总结如下:(1)利用三电极装置模拟了电池环境中溶解气体的还原,精确测定了CO2、O2的还原电位及相应还原产物。(CO2→Li2CO3,2.58 V vs. Li/Li+;(2)O2→Li2O2,2.68 V vs. Li/Li+)(图1、2);(2)根据傅立叶变换红外光谱和核磁共振的结果,结合MD模拟,证实了CO2与TFSI阴离子之间存在强相互作用且在5 m处达到最大,导致5 m中CO2处于一种富集和稳定的状态,不易被吹扫等物理方法清除(图4);(3)在CO2-SIW电解液中,全电池(LiMn2O4-Mo6S8)在低倍率和高倍率下均具有良好的循环稳定性,在0.5 C下进行100次循环后容量保持90 %(初始容量:95.94 mAh/g,100次循环:86.12 mAh/g),在2 C下进行2000次循环后容量保持近70 %(初始容量:83.12 mAh/g,100次循环:57.50 mAh/g)(图5a-d);(4)在CO2-SIW电解液中,全电池(LiMn2O4-Mo6S8)表现出更好的动力学性能。与WIS相比,CO2-SIW不仅具有更好的倍率性能,在15 C时容量保持率高达70 %,而且使用 300 um以上的超厚Mo6S8电极(面载量为 40 mg/cm2)时,依然可以实现90 mAh/g的初始容量,50周之后的保持率高达88 %。它在-40 ℃也能正常工作, 在0.5 C时可以保持70 mAh/g的容量且90周之后无明显衰减趋势(图6);(5)首次在低浓度电解质溶液中形成了稳定的SEI。在CO2-SIW电解液中,全电池循环前和循环后的Nyquist图和XPS光谱结果显示,Li2CO3富集在SEI的外层,LiF富集在SEI的内层(图5e-f)。
另外,高盐浓度LiNO3电解液在水系锂离子电池中已经应用几十年(JES 142,6,1995),同样存在溶解O2和CO2,却未发现有Li2O或Li2O2或Li2CO3的形成,说明除了高盐浓度之外,阴离子化学也是诱导形成SEI膜的关键,与研究结果一致。即溶解CO2与TFSI阴离子间独特的强相互作用使得CO2得以在SIW电解液中以一种富集稳定的状态存在,进而被还原生成SEI起到保护作用。
研究工作首次发现了LiTFSI-CO2之间存在特殊相互作用,这种存在于锂盐-气体-水间的新化学态为实现高电压水系电池提供了全新的界面调控手段,使得开发宽电位水系电解液不在单纯依赖超高盐浓度来实现,为宽电位水系电解液的设计开发提供了新思路。
相关研究成果以Aqueous interphase formed by CO2 brings electrolytes back to salt-in-water regime为题,发表在Nature Chemistry上。研究工作得到国家自然科学基金、物理所、北京凝聚态物理国家研究中心、北京清洁能源前沿研究中心、怀柔清洁能源材料测试诊断与研发平台以及北京材料基因工程高精尖创新中心的支持。

图1.不同气体氛围下WIS电解液中首周放电容量的定量分析。(a)三电极装置工作环境示意图,其中AC代表活性炭;(b)三电极装置下,Mo6S8在不同气体处理后的WIS电解液中的首周放电曲线,其中Mo6S8在有机体系扣式电池中的首周放电曲线用作对比;(c)与Mo6S8在有机体系中的结果相比,不同放电阶段气体还原贡献的容量

图2.三电极装置下,Mo6S8在不同气体氛围下的WIS电解液中所形成的固态电解质界面SEI膜的成分分析。(a)三电极装置下,Mo6S8在不同气体处理后的WIS电解液中的首周放电曲线汇总。XPS及Ar离子刻蚀后的XPS联合TEM的结果来表征不同气体氛围下WIS电解液中循环后的Mo6S8的界面化学及结构;(b)CO2;(c)Ar;(d)O2;(e)Air

图3.5 m LiTFSI溶液的物理化学性质。(a)LiTFSI水溶液电导率和黏度随浓度的变化,正如粉红色阴影所展示的,5 m表现出最高的电导率和最小的黏度;(b)依据1 m,5 m,10 m和15 m的DSC结果给出的LiTFSI-H2O的两相分布相图,5 m具有更低的液相点温度(-31 ℃);(c)由21 m到1 m溶液成本的变化趋势以及M(mol/L)与m(mol/kg)的对应关系;(d)不同气体处理后5 m LiTFSI溶液的CO2含量及pH的对比,control是指没有处理过的5 m LiTFSI溶液,所有样品都测试了三次以求取平均值,相应的标准偏差以误差条的形式给出

图4.CO2气体处理前后CO2与TFSI阴离子的相互作用。(a)CO2处理前后5 m LiTFSI溶液的13C,1H和17O核磁共振谱;(b)不同浓度电解液用CO2气体处理后的红外光谱对比;(c)不同浓度LiTFSI 电解液用CO2气体处理前后19F的NMR化学位移对比;(d)给出了(c)中相应的偏移值;(e)TFSI阴离子中F元素周围电子云密度变化的分子动力学模拟分析(Δρe = ρe,CO2-dealt - ρe,CO2-control);(c-e)的结果显示CO2与TFSI阴离子之间具有较强的相互作用

图5.水系全电池(LiMn2O4/CO2-SIW/Mo6S8)的电化学性能,其中LiMn2O4与Mo6S8的质量比是2:1,比容量的计算是依据负极的质量来计算的。(a)全电池在CO2-SIW电解液以及对比电解液中的首周充放电电压曲线,库伦效率被标注在图内;(b)全电池在0.5 C下的循环性能以及对应的库伦效率(c);(d)全电池在2 C下的长循环性能;(e)CO2-SIW电解液下全电池的电化学阻抗谱及对应的拟合结果;(f)Mo6S8电极循环前后的XPS(C1s和O1s)图谱

图6.CO2-SIW电解液和WIS电解液下LiMn2O4-Mo6S8全电池的动力学性能,LiMn2O4与Mo6S8的质量比为2:1,比容量的计算是依据Mo6S8负极的质量来计算的。厚电极(40 mg/cm2,300 μm)下的。(a)首周充放电曲线以及(b)循环性能;低温(-40 ℃)下的(c)首周充放电曲线以及(d)循环性能
免责声明:本文仅代表作者个人观点,与中关村新型电池技术创新联盟无关。其原创性以及文中陈述文字和内容未经本网证实,对本文以及其中全部或者部分内容、文字的真实性、完整性、及时性本站不作任何保证或承诺,请读者仅作参考,并请自行核实相关内容。
凡本网注明 “来源:XXX(非中关村新型电池技术创新联盟)”的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责。
如因作品内容、版权和其它问题需要同本网联系的,请在一周内进行,以便我们及时处理。电话:010-62899890 邮箱:119@battery100.org

08-25 来源:科技日报

08-20 来源:腾讯汽车

06-09 来源:中国工业报
07-01 来源:中科院大连所

06-07 来源:电池百人会-电池网